ALK基因突变和对应的靶向药物(最全梳理)0 Q7 O8 U2 s6 C/ X& [0 G" |7 o
ALK基因突变和对应的靶向药物4 W2 H& s, h7 W$ G9 F7 x
间变性淋巴瘤激酶(ALK)突变的形式有过量表达、与其他基因形成融合基因,发生点突变等等。ALK基因融合突变是非小细胞肺癌(NSCLC)常见的一种驱动基因,中国非小细胞肺腺癌中ALK融合突变阳性的比例为5.3%,在非小细胞肺腺癌、年轻患者(小于60岁)以及不吸烟的人群中发生率较高,ALK阳性的非小细胞肺癌被认为是一种分子亚型,相对应的靶向药物与EGFR分子亚型完全不同。
# h9 E7 `+ ^, ~ALK融合基因突变主要在肺腺癌里常见,一般肺鳞癌患者ALK融合基因突变概率很低,有报道说1400个肺鳞癌患者里ALK融合基因的发生率为1.3%。考虑到ALK总体突变频率仅有5%,所以对于鳞癌患者也是可以做一下ALK检测的。由于非小细胞肺癌里的驱动基因突变一般是互相排斥的,或者说一山不容二虎,癌细胞也没有必要搞两个驱动突变。有研究说亚裔的EGFR、KRAS野生型的腺癌患者,ALK阳性比例高达30%-42%,因此如果发现EGFR和KRAS是野生型,是更有必要测下ALK基因的。
# G% ^; l2 M/ c# X! H13 k) @! ]- F2 O3 p- y0 z
ALK融合突变的检测+ @, j! G# ~1 h- F: w
 [; I# I) b0 n0 _, u# E6 d/ {! [
[; I# I) b0 n0 _, u# E6 d/ {! [
图1:非小细胞肺癌中ALK的重排形式) A$ [6 N( f1 _8 [1 `/ y( O: J: L
据报道,目前已发现21种EML4-ALK的融合形式,另外ALK还可能与TFG、KIF5B、KLC1、PTPN3、STRN等基因发生融合,因此ALK融合突变的诊断是存在一定难度的。下表是关于ALK融合突变的诊断方法,及其相应的特点。. \, z- y: h* {7 S* P0 G

1 A7 u+ }- T' W+ V表1:ALK基因检测的方法8 R8 {- W N* \0 Q: {; @1 e3 X
需要注意,临床常用的三种方法是FISH、Ventana IHC及RT-PCR,三种方法FISH的灵敏度最低。因此,如果是胸腔积液、细针穿刺取到的细胞学样本做成的蜡块,不建议使用FISH,避免假阴性。另外通过抽血检测循环肿瘤DNA(ctDNA),循环肿瘤细胞(CTC)也正在发展起来。总之在面对ALK检测结果模棱两可的时候,一定要换一个检测方法去验证,也没有哪一种方法灵敏度和特异性都是100%。
) O8 o; e& u |5 h0 S6 a/ K1 j7 }27 T1 R' e3 ]# X4 y5 O3 X/ V3 Y
ALK的靶向药物5 K8 C w: m3 ?
ALK融合突变阳性的患者使用克唑替尼可以获益,克唑替尼具有ALK、c-MET、ROS1三个靶点。克唑替尼治疗ALK阳性的非小细胞肺癌客观缓解率达60%,无进展生存期为8-10个月,显著改善并延长的总生存期。需要注意的是,克唑替尼的赠药政策与其他靶向药物不同,第一年买四个月赠八个月,第二年仍需要买四个月,才能终身获赠,合计下来得几十万,价格相对较高,所以使用之前一定明确是ALK突变才行。
+ q# A8 C0 v t# l

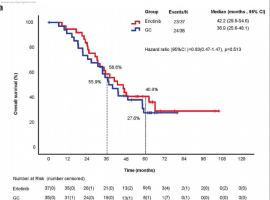
p/ L" e: ~( o4 w/ X: f图2:相比多西他赛、陪美曲塞,ALK阳性的肺癌患者使用克唑替尼获益明显。
: V6 ~& {- d/ E, o7 G; D( g8 n不管如何,靶向药物都有一个短板就是耐药,使用克唑替尼的患者往往在1-2年内出现对克唑替尼的耐药,以中枢神经系统的复发进展较为常见。
3 U* m- f( _5 [6 X

* j: [* `+ e# d: N1 D4 y- L表2:ALK耐药的原因和应对策略7 ]$ k# X. b, H4 K. W% ~
ALK靶向药物 R( k- J, d/ d3 k
1 t3 H" n& i! S1 \. P克唑替尼耐药后,后续还有二代,三代的ALK抑制剂,最近的发现三代ALK抑制剂劳拉替尼(3922)耐药后,患者如果是存在L1198F导致的耐药,可以可以重新用回克唑替尼。几种二代、三代ALK靶向药物的简介如下:1 q! W7 ~7 c+ l1 Q2 d) e
1
+ L: t9 N; X7 i艾乐替尼(Alectinib,代号CH5424802)
+ r' ^/ W9 j1 Y9 M/ @7 o# E2 l效率比克唑替尼强10倍,可对抗大多数的ALK激酶区突变,且对脑病灶控制较好。日本的一项临床研究使用的计量为每次300mg、每日两次,46名患者的43名获得客观缓解(客观缓解率达93.5%),日本已经批准了该药使用,美国FDA也已经批准该药用于克唑替尼治疗后耐药的患者。2016年ASCO会议上报道了一项研究,艾乐替尼一线治疗ALK阳性非小细胞肺癌中的无进展(PFS)显著优于克唑替尼。克唑替尼的中位PFS为10.2个月,而艾乐替尼的中位PFS要大于20.3个月。相比克唑替尼,艾乐替尼使得疾病恶化或死亡风险显著降低66%。
1 A7 |1 f9 I1 m0 S# z) K/ W9 |# g; H2
0 w: u/ P% s5 O( z. ]0 B色瑞替尼(Ceritinib,LDK378)! T7 S4 Z; ~* p- q
对C1156Y具有良好的活性,该药的最大耐受计量为每天750mg,亚裔人的耐受计量可能到不了那么高,又说600mg的。79例克唑替尼耐药的ALK阳性非小细胞肺癌使用该药后,ORR为57%。一项涵盖114名患者的临床表明色瑞替尼的中位PFS为8.6个月。最常见的副作用是恶心、腹泻、呕吐和乏力。有患者反映色瑞替尼的副作用非常大,很少有人耐受,但是如果撑过去了则可能获益期较长。3 Z. Q+ d7 U3 d" M: x- s" K* D
3
7 L/ S& q2 U3 t, n5 L! UBrigatinib(AP26113). P* j( w0 P( F7 m
一种新型的ALK和EGFR双重抑制剂,可强效抑制ALK的L1196M突变和EGFR的T790M突变。2016年ASCO会议上公布的一项研究结果,即将患者1:1随机分为两组,A组患者每天口服Brigatinib药物90mg,B组患者前7天每天口服Brigatinib药物90mg,后面加量到180mg,两组人群的ORR分别为46%/54%,A组有一例证实的完全缓解,B组有五例证实的完全缓解,中位PFS分别为8.8个月/11.1个月。证明了该药良好获益。# ?" l1 ]4 S( a: _
46 P" e3 r. K. H: Y
劳拉替尼(Lorlatinib,PF06463922)
" V& l) _ ^ _( ]该药应该算是第三代ALK抑制剂,可抑制克唑替尼耐药的9种突变,具有较强的血脑屏障透过能力,入脑效果较强,特别适合对其他ALK耐药的晚期NSCLC患者。2016年6月5日,辉瑞在ASCO会议上公布了该药的I/II期临床研究数据,入组的54例患者有41例为ALK阳性,12例为ROS阳性,其中39例有脑转移。该临床试验最终确定的给药方案为每日1次100mg,患者的总应答率为46%,3例实现完全应答,16例实现部分应答,中位PFS为11.4个月,另外还显示出缩小转移性的脑部肿瘤体积的效果。
2 g5 f9 u+ |* V1 v* Z另外还有几个药物如X-396,ASP3026等,其相应的靶点和相关数据见下图,如有兴趣的可以追溯相关参考文献进行延伸阅读。' j& |" \4 e+ z5 ~" F( l" S

' |( Y+ @% L u, l7 h# p图3:ALK新一代抑制剂的特点,最后两列为可以克服的克唑替尼耐药位点,以及无能为力的位点。
a% t, k& L7 h! t' {' B( ?33 r* U2 ~. p" d7 w4 P
HSP90抑制剂与ALK耐药
% ?4 E* L; s& T联合热休克蛋白(HSP90)是一类称为“分子伴侣”的蛋白质,可帮助新合成的蛋白质形成发挥他们特定生物学功能的正确形状。体外细胞系研究发现HSP90抑制剂Ganetespib对ALK阳性的细胞系有活性,且不管是否经过克唑替尼处理都是如此(见下图)。我们可以看到突变的EML4-ALK和HSP90是需要相互结合的。目前有关HSP90抑制剂Ganetespib的临床试验正在进行中。: _ u: R+ U2 u) j. A; I0 y
另一种HSP90抑制剂是AUY922,目前正在进行ALK阳性的NSCLC的II期临床试验,每周计量70mg/平米。疾病控制率为59%(未经克唑替尼治疗的控制率为100%,克唑替尼耐药组的为36%)。
' A4 L- M2 P5 b

8 y5 r! c/ P, |3 k( ~. Z5 G5 j$ D' d% t图4:ALK阳性NSCLC的靶向治疗机制 I. ~ Z" y1 S3 c$ c% b$ D
4
' p4 @ c7 k% O9 FEGFR和ALK双突变患者1 t+ m2 |$ e ]7 r. H) K3 g) `9 Y
普遍认为,ALK和EGFR基因是互斥的,因为肿瘤其实没有必要制造两个驱动基因。但是对于后期经过多种治疗后,反复耐药的患者。EGFR和ALK共存的概率也不容忽视。对于这一部分患者,联合使用EGFR和ALK的抑制剂较好,比单独使用一种起到更好的控制作用,但是联合治疗副作用会较大,也需要看患者的耐受情况。目前关于两类靶向药物联用效果、副作用等还缺乏数据。当然这部分患者可以考虑下Brigatinib(AP26113),该药是ALK和EGFR双靶点的抑制剂,可以考虑参加入组试验等。但问题是该药可以抑制EGFR和ALK守门员突变(T790M,L1196M),如最开始就使用Brigatinib,这可能是把最后一张牌给打了。
, M0 o/ H9 {+ i( v) w" D2 B笔者曾见过一个原发性双突变的患者,同时具有EGFR和ALK阳性,该患者的治疗情况也将及时追踪,后续再和大家呈报。; f, R% P9 S" `
5
1 h* @0 d# \& [) _什么时候停药' @4 m' k6 E8 f- h2 a9 v% S
即便是出现局部进展,也不是立刻停止克唑替尼等靶向药物的理由,因为可能还有很多癌细胞被药物所抑制。立即停止靶向药物,会导致肿瘤的爆发性进展。
/ v/ ?8 u: A; ?8 s+ x3 {' I一项关于使用克唑替尼抑制ALK阳性非小细胞肺癌的研究发现,克唑替尼治疗进展后,继续使用克唑替尼相比停止克唑替尼的患者获益更好。6个月总生存率为76.3% vs 31.2%,1年总生存率为64.7% vs 32.9%,OS为16.4个月 vs 3.9个月。
5 T) h# x6 \8 M' l也有部分患者使用克唑替尼后,肿瘤病灶快速消失掉了,于是患者把药给停止了,后来导致报复性的复发,有些患者再用克唑替尼也控制不了,即是一种脱靶效应,虽然这其中的机理不知道,但是确实是存在的。这些现象是某些患者的血泪教训,虽然没有临床数据,但值得警惕。因为CT看不到肿瘤了,不代表肿瘤细胞完全被杀灭干净了。停药是一个非常谨慎的事情,望广大患者和家属慎之再慎。
+ t( O! }% d% m6/ V* O3 {. E; ^( w' d8 w
完美的闭环?
& L4 m! S. y: Y$ e) i* C劳拉替尼(Lorlatinib,PF06463922)被作为ALK靶点的最后一张王牌,因为克唑替尼耐药的所有位点该药似乎都能克服。直到出现了L1198F。新英格兰医学杂志报道了一名患者治疗经过(见下图)。该患者首先使用克唑替尼,耐药后检测发现了C1156Y,但是这个患者对二代ALK抑制剂没有应答,不得已使用劳拉替尼,但是后面新出现的L1198F导致对劳拉替尼耐药,看似山穷水尽,却不曾想L1198F突变导致了与克唑替尼结合更好,逆转了C1156Y的作用,患者对克唑替尼重新复敏。
' ]5 i; r, H0 \# ~
 & h! d& v6 [" u; t: h8 [% L5 B
& h! d& v6 [" u; t: h8 [% L5 B
图5:L1198F导致的劳拉替尼耐药对克唑替尼重新复敏
6 ^6 x& _) `/ k9 L8 z! ?, \这是一个非常有意思的发现,即劳拉替尼这个药物也不是最后的一张牌,这个药物耐药了,也许之前被放弃的药物仍可以有效。所以也有说法ALK是一种钻石突变。但需要谨慎的是不是每一个劳拉替尼耐药的患者都这样。我们从图示看出患者同时存在C1156Y和L1198F突变,才造成了这种逆转。也许其他的突变位点如L1196M等和L1198F就没有这种协同效果。或者还有可能劳拉替尼耐药的原因是其他的旁路激活,如KRAS或EGFR等,具体的还是可以根据基因检测结果来谨慎分析和对待,不过现在的测序技术抽血测ctDNA检测ALK的这些激酶突变总可能会有阴性,ArmsPCR等检测方法也还没有相应的产品,暂时只能尽量地取耐药后的新发组织样本,进行二代测序检测。
# C" @) X4 T7 l% U" @' A参考文献:/ d3 T4 w3 D l) x' ?9 E
1、冯勤 杨欣 林冬梅,ALK阳性非小细胞肺癌的诊断,中国肺癌杂志2 0 1 5年2月第1 8卷第2期。) W0 e, k9 @2 r- a# n$ `
2、蒋涛 周彩存,中国肺癌杂志2015年2月第1 8卷第2期' w ~$ i! W% z: P! [, c6 @& F( [, O
3、Alice T, J Clin Oncol. 2013 Mar 10; 31(8): 1105–1111
- k$ n+ h& S2 e2 O, G! g& f1 M$ u; {2 ~6 N4、Clin Cancer Res. 2014 March 1; 20(5): 1204–1211
& L. Z; |# B) [/ u6 X* e5、Ou SH , et al. Ann Oncol, 2014, 25(2): 415-422.
4 O6 ^1 ]& \) E# M5 ?* t/ @6、Zhang I,Lancet Oncol. 2015 Oct;16(13): e510-21' S, u% Y [; q
7、Ther Adv Med Oncol. 2016 Jan;8(1):32-476 q% s- z0 Z* P/ p
8、Shaw AT et al, N Engl J Med. 2016 Jan 7;374(1):54-61 |
 非小细胞肺腺癌四期骨转脑转肝转
非小细胞肺腺癌四期骨转脑转肝转,基因突变KRAS PG-12D,四线治疗安罗替尼耐药,目前
非小细胞肺腺癌四期骨转脑转肝转
非小细胞肺腺癌四期骨转脑转肝转,基因突变KRAS PG-12D,四线治疗安罗替尼耐药,目前
 御风而行 因爱共赢——第一届肺癌病
作者:Tony
近年来,随着靶向、免疫、ADC等药物的蓬勃发展,晚期肺癌已逐渐转变成为可
御风而行 因爱共赢——第一届肺癌病
作者:Tony
近年来,随着靶向、免疫、ADC等药物的蓬勃发展,晚期肺癌已逐渐转变成为可
 母亲肺腺癌,9年靶向耐药后,如何选
2017年5月,因持续干咳入院检查,肺部弥漫型结节,支气管镜确诊肺腺癌,四期。基因检
母亲肺腺癌,9年靶向耐药后,如何选
2017年5月,因持续干咳入院检查,肺部弥漫型结节,支气管镜确诊肺腺癌,四期。基因检
 肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
 肺癌术前新辅助治疗,靶向治疗能否替
作者:闵
手术是广大病友眼中治愈癌症的神兵利器,可惜有一部分病友尽管处于中早期,
肺癌术前新辅助治疗,靶向治疗能否替
作者:闵
手术是广大病友眼中治愈癌症的神兵利器,可惜有一部分病友尽管处于中早期,












 显身卡
显身卡














